安全药物的选择性在靶点验证过程中是非常重要的。选择性对于药物设计增添了不少的复杂性,因为它设计到多重结合亲和力的优化过程。从计算角度来讲,结合选择性预测是具有挑战性的,通用的方法仍无法使用。基于化学途径的绝对结合自由能计算为亲和力预测提供了框架。本文作者利用三个溴结构域抑制剂,评价了基于分子动力学模拟计算得到的自由能,并和等温滴定量热法数据进行了比较。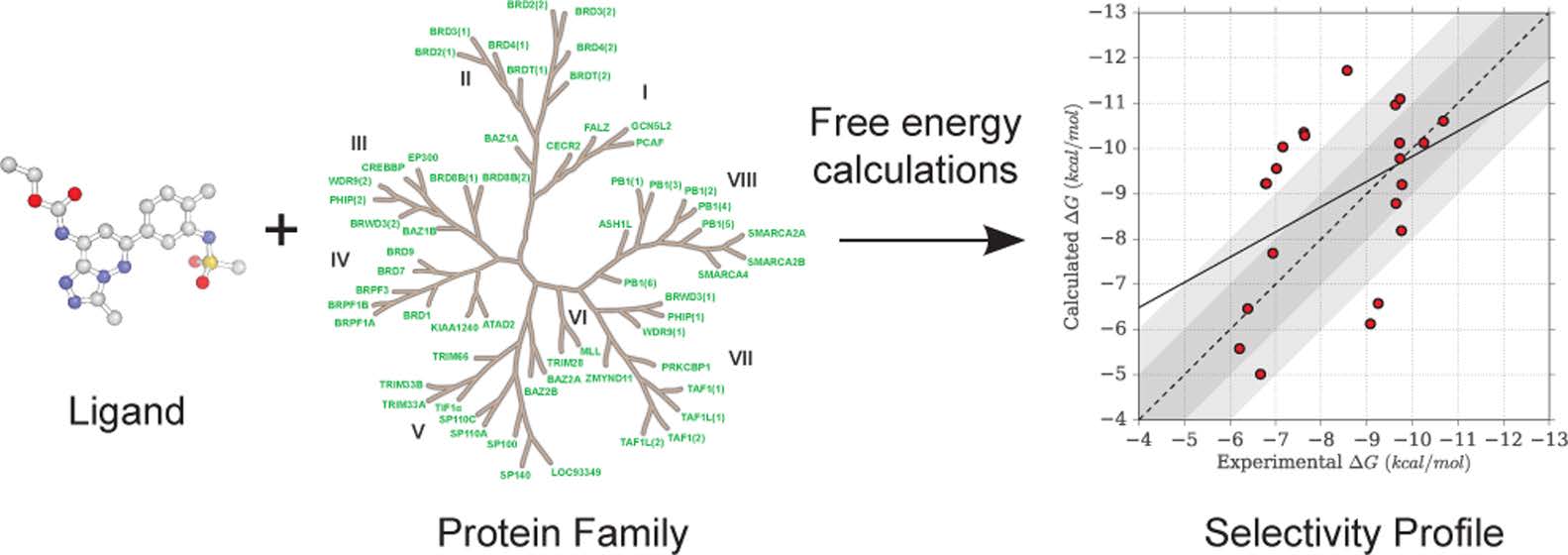
结合选择性是安全治疗中最重要的需求。预定靶点和相似蛋白间结合亲和力的差异可以用来避免不需要的副作用。设计一个选择化合物工具是一个困难的任务,尤其当蛋白家族具有保守的折叠区和相似的结合口袋残基时。蛋白激酶家族就是个很好的例子。溴结构域可以特定的识别ε-N-lysine acetylation motifs。46种人体细胞核和胞浆蛋白中发现了61种溴结构域蛋白。根据序列和结构的相似性,它们被划分为八个家族。虽然它们结构具有多样性,但它们都具有一个保守的折叠区。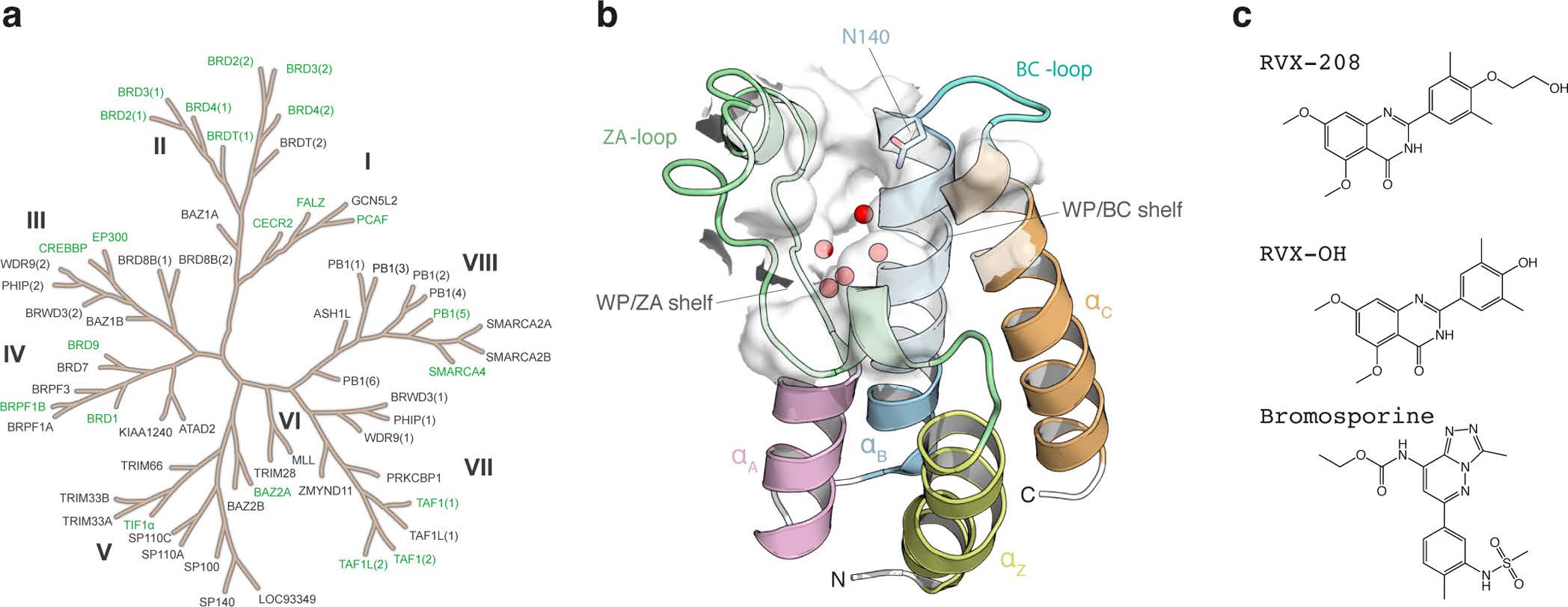
选择性为合理设计新药增添了复杂性。目前,多种策略已经被用于选择性设计,如形状和静电互补,构象选择,水转换以及别构结合。然而,选择性仍然是药物设计过程中的一个挑战。基于分子动力学模拟的结合自由能计算提供了另一种方来来预测结合亲和力。目前,绝对结合自由能(ABFE)方法已经被用来比较不同配体与任意靶点蛋白之间的亲和力。本文作者计算了36个复合物之间的亲和力,包括22个溴结构域蛋白和3个配体。
最后,作者通过ABFE计算发现,ABFE计算在先导物发现和先导物开发过程中具有较高的潜力。分子对接和ABFE可以被用来评价配体和不同蛋白之间结合可能性。
实验方法:
1.molecular docking(MOE).
2.Molecular Dynamics(Gromacs 5.0.4).
3.Free Energy Calculations.
4.Data Analysis.
使用到的包: python package pymbar and alchemical analysis tool.
Reference
Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations.链接:http://pubs.acs.org/doi/abs/10.1021/jacs.6b11467